There is a limited amount of data out there that suggests that many researchers overpromise on the planned sample size and completion date and underdeliver. For example,
- “Globally, more than 80 per cent of clinical trials fail to enrol on time, and this recruitment problem is extremely costly for drug companies, contributing to 85-95 per cent of the lost days in a clinical trial.” Source
I attended a web seminar about optimizing clinical trial enrolment, and they offered some additional evidence of the problem
- More than 90% of clinical trial sites delay enrollment – CenterWatch
- 72% of studies are delayed by more than a month – CISCRP
- Out-of-pocket costs are 1.2M on average per study for each month delayed – Top 10 Pharma Company report
- Delays in studies can cost life-sciences companies at least 800,000 a day in lost sales for a niche medication and $5.4 million a day for a blockbuster – McKinsey
About a year ago, I received a small grant to study the proportion of studies at Children’s Mercy Hospital (CMH) that failed to meet the proposed completion deadlines, that failed to recruit the promised number of patients or both. Here is a brief summary of these results.
Records were reviewed for a stratified random sample of 125 IRB approved studies requiring full review which produced final reports between January 2001 and December 2005. Studies requiring full review were to a large extent studies that were prospective. Studies were stratified by year of completion. We excluded any studies from the Children’s Oncology Group (COG). Childhood cancer is a thankfully rare disease, so the typical COG protocol would enroll only 1 or 2 patients at each site. We also excluded any retrospective studies or any studies not involving humans. If the study were part of a multi-center trial, we calculated the planned and actual sample sizes from CMH only.
In addition to this random sample, a 100% census of all internally funded studies (KBR studies) was examined.
For each study, the final report, any interim reports, and the original protocol submission were reviewed. The variables recorded were
- planned study start date,
- planned study end date,
- actual study start date,
- actual study end date,
- planned sample size,
- actual sample size.
If certain dates or sample sizes could be determined from the study, a code of U was entered. The primary goal of this research was to estimate
- the proportion of studies which lasted longer than planned,
- the proportion of studies which recruited fewer subjects than planned,
- the average size of these deviations.
A secondary objective was to measure the proportion of times that information about planned start and end dates were not included in the file. Finally, we wanted to see if certain features of the studies
- External sponsor (yes / no),
- Study coordinator (yes / no),
- Consent required (yes / no), and
- Randomization used (yes / no)
were associated with any of these measures.
For the sample of 125 IRB approved studies, we were only able to determine the performance against the proposed deadlines in 9 studies. Information was not supplied or was ambiguous on the planned start date (n=2), the planned end date (n=87), or both (n=25)), allowing the calculation of the planned duration in only 11 (9%) of the studies. The CMH IRB does not require applicants to include either date in their protocol submission, which is a major failing. For these 114 studies, the IRB was essentially signing a blank check and implied that approval was not contingent on the timely conduct of the study. Of the remaining 11 studies, one did not have an actual start date listed and one did not have an actual end date listed.
For the remaining 9 studies, the average planned duration was 15 months (range 4.6 to 36 months). In seven of the studies, the actual duration was longer than the planned duration and in two studies it was shorter. The average change in the duration was 191% (range 87% to 386%).
The information on planned and actual sample sizes was more complete. There were 19 studies where the planned sample size could not be determined and 1 study where the actual sample size could not be determined. This allowed us to calculate the sample size shortfall or surplus in 105 (84%) of the studies.
For these studies, the average planned sample size was 51 (range 1 to 830). In 59 (56%) of those studies, the researchers recruited fewer patients than planned. There were 9 studies (9%) that failed to recruit any patients at all. Of the remained 46 studies, 23 (22%) recruited the exact number of planned subjects and 23 (22%) recruited more than the planned number of subjects. The average study reached 75% of the planned sample size (range 0% to 200%).
Among the 17 KBR funded studies, the results were similar. Only 3 (18%) provided sufficient information to compare the planned duration to the actual duration. The planned durations were 8, 9, and 12 months, while the planned durations were 7, 35, and 12 months respectively. There were 14 studies with sufficient information to compare the planned and actual sample sizes. There were 7 studies (50%) that recruited fewer subjects than planned, though no studies recruited zero patients. There were 3 studies (21%) that recruited exactly the planned number of patients and 4 studies (29%) that recruited more patients than planned. The average study reached 84% of the planned sample size (range 30% to 133%).
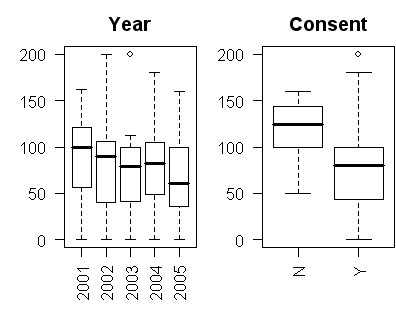
There was insufficient data to examine trends and patterns in sample size shortfalls among the KBR funded studies. In the sample of IRB approved studies, there was a decline in the percentage over time. Studies requiring informed consent had much more trouble reaching their target sample size than studies where the informed consent requirement was waived. There were only 7 studies, however, where the informed consent requirement was waived. There were no other factors which had a major influence on the ratio of actual to planned subjects.
Only 24 studies (19%) commented on subject shortfalls or delays. The most common reason cited (n=16) was reluctance of patients/parents to enroll in the study. Less commonly cited was loss of external support (n=3), stringent inclusion criteria (n=2), time constraints (n=1) or multiple reasons (n=2). There was insufficient data to allow further analysis of these reasons.
Conclusions: The current IRB reporting mechanisms at CMH do not require researchers to report the planned duration of their research trials, nor do they require researchers to comment on any delays in their trials. I suspect that this is similar for many other IRBs. This represents a serious failing on the part of the IRBs to monitor the progress of these trials. While small delays are tolerable, the scientific validity of a trial comes into question if the proposed time frame or the actual time frame of the proposed research extends beyond a reasonable limit. Research delayed is research denied.
About half of the IRB approved studies failed to enroll the promised number of patients. The average study fell short of the target by 25%.
Only a small number of researchers provided comments about shortfalls or delays. The predominant reason provided was reluctance of patients to volunteer for the study.
You can find an earlier version of this page on my original website.